
 A rhodium-based catalyst reverses regioselectivity in the production of hydrazones
A rhodium-based catalyst reverses regioselectivity in the production of hydrazones




.jpg)







Work in the authors’ laboratory on toxins and pain is supported by grants from the Australian Research Council (DP1093115) and the National Health and Medical Research Council (631457, 1010552, and 1026501.
The black mamba snake here illustrates the structure of mambaglin-1, a pain-relieving peptide found in its venom. Thick gray lines lines represent the four disulfide bonds linking Cys1–3, 2–4, 5–6, and 7–8. The N-terminus of the 57 amino acid peptide is at the head of the snake and the C-terminus at the tail. The peptide has potential as a pharmacological probe or drug lead. Image design by David Craik and drawing by Peta Harvey, University of Queensland.
Mambalgins Mambalgins classified as being part of the family of three-finger toxins. There are two isoforms of mambalgin which have been given the names of mambalgin-1 and mambalgin-2. Both of these isopeptides are made of a 57 aminoacidic chain with 8 residues of cysteine. These two isoforms differ only in the residue located in the fourth position of the chain.
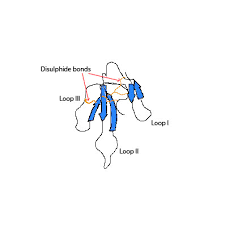
Mambalgin-1 presents a three-dimensional structure which consists of 3 loops emerging from the nucleus of this protein. A triple chain with antiparallel β-sheets connects loops II and III, and a double chain, also formed by antiparallel β-sheets, allows the formation and bonding of loop I. Moreover, the protein presents a concave area, which is typical of neurotoxins, stabilized by four disulphide bonds.
Mambalgins also show a high electrostatic potential which is necessary for the bounding with the ASIC ionic channels, http://flipper.diff.org/app/items/5368 which are negatively charged.
These toxins have been proved to have a strong analgesic effect in both central and peripheral nerves, being able to be as potent as morphine but better because they cause less tolerance and no respiratory distress. http://www.ncbi.nlm.nih.gov/pubmed/?term=mambalgine
While morphine acts on the opioid pathway of the brain causing addiction, headaches, difficulty thinking and vomiting, mambalgins avoid pain using a completely different route, which is potentially capable of causing fewer side effects. Mambalgins have been found to take away pain by inhibiting acid-sensing ion channels (ASIC) in the peripheral and central nervous system.
When an external pain stimulus is received by the body, our damaged cells release an inflammatory soupcontaining ions and other chemicals. The ions released by the damaged cells are detected by the ASICchannels, which open up due to a change in the pH levels that the ions cause. As the protein ion channels open, they trigger the electric impulse sent to the brain telling him that the body is suffering.
What mambalgins do in order to avoid pain is that they bind to these ASIC channels and prevent them from opening, thus causing the body not to send pain messages to the brain and acting as a powerful analgesic.
Finally it is important to remember that mambalgins do not block all ion channels, because these kinds of toxins are normally highly specific. Researchers have found that mambalgins have a potent, rapid and reversible effect in homomeric ASIC1 and heteromeric ASIC1a+ASIC2a or ASIC1a+ASIC2b channels, which are the entire ASIC channel subtypes found in the central nervous system. On the other hand, they have no effect on ASIC2a, ASIC3, ASIC1a+ASIC3 and ASIC1b+ASIC3 channels.
Bringing mambalgins to a practical use, these peptides could be useful in the analgesic treatments of patients with chronic respiratory diseases, given that their effect seems not to affect the respiratory system, in contrast to morphine which can cause respiratory distress. In addition, this new potential drug seems to deal with the problem of tolerance using morphine, in other words, needing an increasingly higher dose each time to obtain the same effect. This would be of great use for the chronically ill, as they could take the analgesic as many times as they wanted without becoming addicts or dependant on the drug.


Mambalgins Mambalgins classified as being part of the family of three-finger toxins. There are two isoforms of mambalgin which have been given the names of mambalgin-1 and mambalgin-2. Both of these isopeptides are made of a 57 aminoacidic chain with 8 residues of cysteine. These two isoforms differ only in the residue located in the fourth position of the chain.
Mambalgin-1 presents a three-dimensional structure which consists of 3 loops emerging from the nucleus of this protein. A triple chain with antiparallel β-sheets connects loops II and III, and a double chain, also formed by antiparallel β-sheets, allows the formation and bonding of loop I. Moreover, the protein presents a concave area, which is typical of neurotoxins, stabilized by four disulphide bonds.
Mambalgins also show a high electrostatic potential which is necessary for the bounding with the ASIC ionic channels, http://flipper.diff.org/app/items/5368 which are negatively charged.
These toxins have been proved to have a strong analgesic effect in both central and peripheral nerves, being able to be as potent as morphine but better because they cause less tolerance and no respiratory distress. http://www.ncbi.nlm.nih.gov/pubmed/?term=mambalgine
While morphine acts on the opioid pathway of the brain causing addiction, headaches, difficulty thinking and vomiting, mambalgins avoid pain using a completely different route, which is potentially capable of causing fewer side effects. Mambalgins have been found to take away pain by inhibiting acid-sensing ion channels (ASIC) in the peripheral and central nervous system.
When an external pain stimulus is received by the body, our damaged cells release an inflammatory soupcontaining ions and other chemicals. The ions released by the damaged cells are detected by the ASICchannels, which open up due to a change in the pH levels that the ions cause. As the protein ion channels open, they trigger the electric impulse sent to the brain telling him that the body is suffering.
What mambalgins do in order to avoid pain is that they bind to these ASIC channels and prevent them from opening, thus causing the body not to send pain messages to the brain and acting as a powerful analgesic.
Finally it is important to remember that mambalgins do not block all ion channels, because these kinds of toxins are normally highly specific. Researchers have found that mambalgins have a potent, rapid and reversible effect in homomeric ASIC1 and heteromeric ASIC1a+ASIC2a or ASIC1a+ASIC2b channels, which are the entire ASIC channel subtypes found in the central nervous system. On the other hand, they have no effect on ASIC2a, ASIC3, ASIC1a+ASIC3 and ASIC1b+ASIC3 channels.
Bringing mambalgins to a practical use, these peptides could be useful in the analgesic treatments of patients with chronic respiratory diseases, given that their effect seems not to affect the respiratory system, in contrast to morphine which can cause respiratory distress. In addition, this new potential drug seems to deal with the problem of tolerance using morphine, in other words, needing an increasingly higher dose each time to obtain the same effect. This would be of great use for the chronically ill, as they could take the analgesic as many times as they wanted without becoming addicts or dependant on the drug.

Image of black mamba snake courtesy of Wikimedia Commons/Bill Love/Blue Chameleon Ventures
A bite from the black mamba snake (Dendroaspis polylepis) can kill an adult human within 20 minutes. But mixed in with that toxic venom is a new natural class of compound that could be used to help develop new painkillers.
Named “mambalgins,” these peptides block acute and inflammatory pain in mice as well as morphine does, according to a new study.
Researchers, led by Sylvie Diochot, of the Institute of Molecular and Cellular Pharmacology at Nice University, Sophia Antipolis in France, purified the peptides from the venom and profiled the compounds’ structure. They then were able to test the mambalgins in strains of mice with various genetic tweaks to their pain pathways. Diochot and her colleagues determined that the mambalgins work by blocking an as-yet untargeted set of neurological ion channels associated with pain signals. The findings were published online October 3 in Nature (Scientific American is part of Nature Publishing Group).
As a bonus, mambalgins did not have the risky side effect of respiratory depression that morphine does. And the mice developed less tolerance to them over time than is typical with morphine.

Image of black mamba's black mouth courtesy of Wikimedia Commons/Tad Arensmeier
Experimenting with the newfound compounds should also help researchers learn more about the mechanisms that drive pain. As the researchers noted in their paper, “It is essential to understand pain better to develop new analgesics. The black mamba peptides discovered here have the potential to address both of these aims.”
Venoms from plenty of other species of animals, including spiders, scorpions, ants and even snails, have also been studied for their analgesic potential.
Just don’t try extracting any of this venom in the wild. There is antivenom for the black mamba snake’s bite, but it is not always available, and without it, the bites are usually fatal. These snakes can move along at speeds up to about 20 kilometers per hour and grow to up to 4.4 meters in length.The African black mamba’s bite has been called “the kiss of death”—and for good reason. It has been estimated that just 10 to 15 mg of venom from the world’s deadliest snake, triggering severe pain, vomiting, shock, and limb paralysis, can kill a person within hours.


Yet within the black mamba’s venomous brew of toxins, a French research team has discovered a small fraction of peptides with surprising properties. Called “mambalgins,” they are not toxic, and, at least in mice, they produce a potent analgesia that equals the pain-relieving effect of morphine—but with fewer adverse effects.
Eric Lingueglia and his colleagues at the Institut de Pharmacologie Moléculaire et Cellulaire in Valbonne, France, including Sylvia Diochot—an engineer specializing in animal venoms and toxins—and Anne Baron, reported the findings in the October 3 Nature.
The new study shows the mambalgins inhibit the activity of multiple acid-sensing ion channels (ASICs), which are major players in pain pathways responding to tissue acidification—a common feature of many pain-generating conditions including acute heat pain and inflammation. The peptides suppress ASIC channel activity both in central neurons and in peripheral nociceptors.
In addition to their potential for yielding novel painkillers with superior side effect profiles, the mambalgins are already revealing unappreciated functions of ASIC channels and new therapeutic targets.
ASICs, first cloned in the 1990s, are cationic channels activated by extracellular protons. They are expressed both in the central nervous system and in peripheral nerves. In rodents, six types are known, arising from four genes—ASIC1-4. Four subtypes are splice variants: ASIC1a, 1b, 2a, and 2b. ASICs can be homomeric—containing one channel type—or heteromeric, made up of different combinations of variants. The roles of ASIC types and subtypes in pain sensation are being explored by the Lingueglia group and others, using snake, spider, and other venoms to both activate and inhibit them.
“Mambalgins have the unique property of being potent, rapid, and reversible inhibitors of recombinant homomeric ASIC1a and heteromeric ASIC1a/2a or ASIC1a/2b channels—that is, all the ASIC subtypes expressed in the central nervous system,” the authors note. Mambalgins had no effect on ASIC2a, ASIC3, ASIC1a/3, and ASIC1b/3 channels.
The functional studies were done in mice, but the investigators showed that the peptides inhibited human ASICs in vitro. “We don’t know if they will have the same effects in humans,” says Lingueglia, “but we are confident, because the peptides act on the human channels, and most of the pain pathways we studied are highly conserved between mice and humans.” Lingueglia reported the peptides have been licensed to a pharmaceutical company in Valbonne for development of human applications.
Opioid-independent actions
The Lingueglia group previously used two other venom toxins, PcTx1 (from tarantula) and APETx2 (from sea anemone), to functionally define certain ASIC channels. Now, in a screen for additional ASIC-blockers, the team identified black mamba venom as a potent, reversible inhibitor of ASIC1a expressed in Xenopusoocytes. Two active fractions were collected—isopeptides they named mambalgin-1 and mambalgin-2. They belong to the class of snake venoms termed “three-finger “ peptides, reflecting their distinctive structure.
When injected intrathecally, mambalgins produced strong analgesia against heat pain as assessed with the tail-flick and paw-flick tests, but the effect disappeared in ASIC1a knockout mice, demonstrating the essential involvement of ASIC1a-containing channels. The effect was as potent as that of morphine, but was not reduced by naloxone, indicating that the inhibited channels were independent of opioid pathways. Moreover, in repeated administration, morphine was associated with tolerance such that after five days, its analgesic powers had all but vanished, while mambalgin-1 continued to be effective—additional evidence of a non-opioid mechanism of action.
In experiments looking at peripheral action, injections of mambalgin-1 into mouse paws reduced acute pain and reversed or prevented inflammatory hyperalgesia created by intraplantar injection of carrageenan. Curiously, the researchers found, mambalgin-1 continued to produce analgesia in peripheral pain pathways in the ASIC1a knockouts. If ASIC1a was not responsible, the scientists wondered, what was? Peripheral nociceptors express both ASIC1a and ASIC1b, but the latter’s role in pain was not known. Mambalgin-1 blocks both ASIC1a and ASIC1b in dorsal root ganglion neurons, the scientists noted; therefore, analgesia induced by mambalgin-1 in the ASIC1a knockouts suggests a critical role of ASIC1b in pain sensation. To prove that, the researchers knocked down ASIC1b, and showed that loss of the channel mimicked the effect of mambalgin to reduce pain in the mice.
“ASIC1b at the periphery—nociceptors—was never associated with any type of pain before,” notes Lingueglia. Also, he says, “the heteromeric channel made of ASIC1a+ASIC2a subunits in central neurons was also not known to be involved in pain before this work.”
“Our results indicate that mambalgins have analgesic effects by targeting both primary nociceptors and central neurons, but through different ASIC subtypes,” the authors say.
The effects of mambalgins stand in contrast to those of the tarantula PcTx1 toxin, which also induces analgesia via inhibition of central ASIC1a, but in an opioid-dependent manner (Mazzuca et al., 2007). The mambalgin experiments, together with these previous findings, indicate that “different pathways involving different subtypes of ASIC channels can lead to different types of central analgesia (opioid sensitive or insensitive),” the authors note.
Mambalgins create analgesia by inhibiting different ASIC channels in the central nervous system and peripheral nociceptors. Unlike the ASIC1a-targeted spider toxin PcTx1, the central actions of mambalgins do not involve endogenous opioid pathways. Image: Nature.