ARKIVOC
2014 Part (i): Special Issue 'Reviews and Accounts', PG 1-20
Recent advances on diversity oriented heterocycle synthesis via multicomponent tandem reactions based on A3 coupling (14-8183LR) [pp. 1-20]
Yunyun Liu, a,b
a Key Laboratory of Functional Small Organic Molecule, Ministry of Education,
Jiangxi Normal University, Nanchang 330022, P. R. China
b College of Chemistry and Chemical Engineering, Jiangxi Normal University,
Nanchang 330022, P. R. China
Full Text: PDF (235K)http://www.arkat-usa.org/get-file/48824/
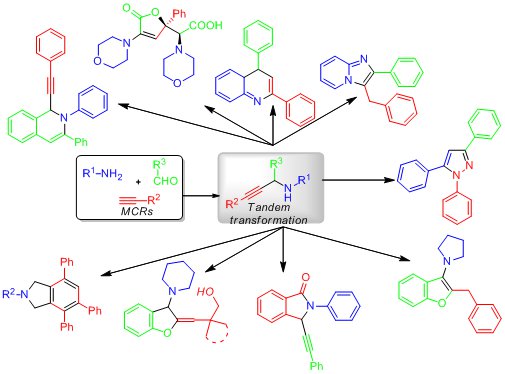
A3 coupling reactions are the reactions between aldehydes, amines and alkynes, which yield
propargylamine derivatives under various catalyst conditions. By making use of the versatile
reactivity of propargylamines, tandem reactions initiated by the functional group(s) in the in situ
generated propargylamines constitute one of the most important applications of A3
couplings.
These tandem reactions are especially useful for the synthesis of heterocyclic compounds. In this
review, the progress on multicomponent tandem reactions based on A3
coupling is summarized.
Conclusions and Outlook
During the last decade, A3
coupling reaction has evolved to a classical three-component protocol
for accessing various propargylamines. Numerous papers have been published on the
investigation of this synthetic method and spectacular advances on A3
coupling reactions have
been witnessed in terms of green catalyst system, asymmetric catalysis etc. which also promoted
this coupling protocol as the most preferred option for propargylamine synthesis. From the
perspective of application, the propargylamines possessed broad spectrum of diversity andreactivity, and these compounds could serve as main building blocks in the synthesis of many
organic small molecules. From the perspective of atom economics, devising tandem reactions
based on key transformation of A3
coupling for the synthesis of more complex and structurally
diverse heterocyclic products in one-pot represent a promising direction in modern organic
synthesis. As introduced in the contents, many elegant results have already been reported on this
area. On the other hand, at current state, this kind of tandem reactions were mainly performed by
using the second functional group in aldehyde, amine or alkyne to initiate subsequent
transformations on propargylamine intermediates, although some reactions using additional
components such as carbon dioxide to design tandem synthesis of heterocyclic products have
also been reported, this kind of examples are still rather rare. Thus, deeper and broader explore is
still demanding since using additional substrates for reactions is theoretically able to provide
considerably higher diversity both in reactions and corresponding products. In addition, versions
of asymmetric catalysis on traditional A3
coupling have already been accomplished with nice
results, while asymmetric catalysis protocols of A3
coupling-based tandem synthesis of
heterocycles kept unexplored, more systematic and advanced approaches of asymmetrical
catalysis on these tandem reactions are expected in future.

CHINA FLAG

BEIJING




.jpg)


 The addition of malonate esters to α,β-unsaturated
The addition of malonate esters to α,β-unsaturated  Asymmetric conjugate addition reactions of alkenylaluminums to
enones
Asymmetric conjugate addition reactions of alkenylaluminums to
enones