Abstract
A mild and green photo-organocatalytic protocol for the highly efficient acetalization of aldehydes has been developed. Utilizing thioxanthenone as the photocatalyst and inexpensive household lamps as the light source, a variety of aromatic and aliphatic aldehydes have been converted into acyclic and cyclic acetals in high yields. The reaction mechanism was extensively studied
Photo-organocatalytic synthesis of acetals from aldehydes
Author affiliations
.////////////////
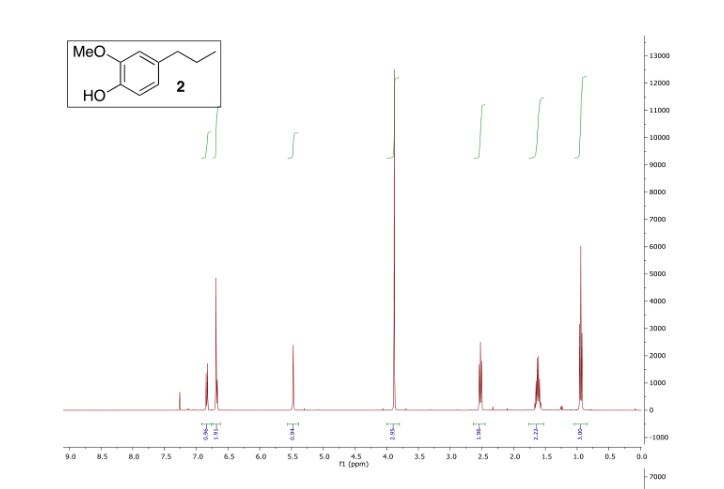
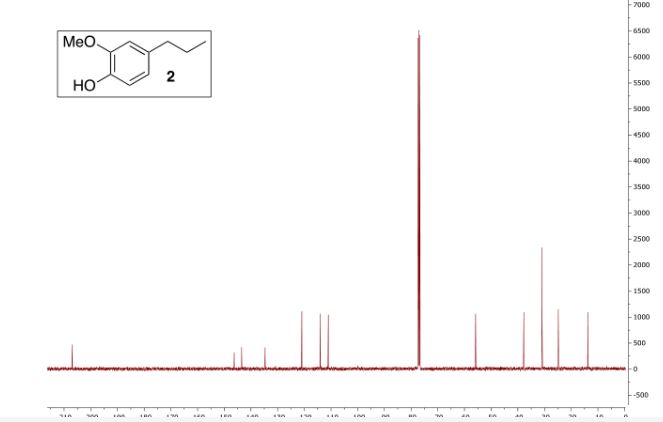
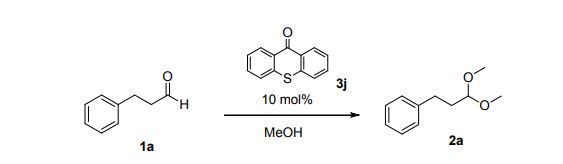
(3,3-Dimethoxypropyl)benzene (2a)6
Colorless oil; 95% yield; 1H NMR (200 MHz, CDCl3) δ: 7.33-7.18 (5H, m, ArH), 4.37 (1H, t, J = 5.8 Hz, OCH), 3.33 (6H, s, 2 x OCH3), 2.68 (2H, t, J = 7.6 Hz, CH2), 1.98- 1.87 (2H, m, CH2); 13C NMR (50 MHz, CDCl3) δ: 141.8, 128.4, 125.9, 103.7, 52.8, 34.0, 30.8; MS (ESI) m/z 181 [M+H]+ .
6. Q. Zhou, T. Jia. X.-X. Li, L. Zhou, C.-J. Li, Y. S. Feng, Synth. Commun., 2018, 48, 1068.
Colorless oil; 95% yield; 1H NMR (200 MHz, CDCl3) δ: 7.33-7.18 (5H, m, ArH), 4.37 (1H, t, J = 5.8 Hz, OCH), 3.33 (6H, s, 2 x OCH3), 2.68 (2H, t, J = 7.6 Hz, CH2), 1.98- 1.87 (2H, m, CH2); 13C NMR (50 MHz, CDCl3) δ: 141.8, 128.4, 125.9, 103.7, 52.8, 34.0, 30.8; MS (ESI) m/z 181 [M+H]+ .
6. Q. Zhou, T. Jia. X.-X. Li, L. Zhou, C.-J. Li, Y. S. Feng, Synth. Commun., 2018, 48, 1068.